CREATING ENZYMES BY DESIGN AND EVOLUTION
Dr. H. Adrian Bunzel
ORCID: 0000-0001-6427-368X
Honorary Senior Research Associate
adrian.bunzel@bristol.ac.uk
+44 (0) 117 331 2172
Group of Ross Anderson
Group of Adrian Mulholland
C101D – Biomedical Sciences Building
University Walk, Clifton BS8 1TD

Lecture: Directed Evolution
Enzymes, designed by Nature over billions of years of evolution, accelerate their reactions with unequaled efficiency. Mimicking that process, enzymes can be created by computational design and directed evolution. We tailor (1) artificial photocatalytic enzymes by design of photosensitizer binding sites into existing biocatalysts. Furthermore, we use experimental and computational methods to dissect the dynamic and electrostatic origins of catalysis (2) in designer enzymes.
(1) Artificial Photocatalytic Enzymes
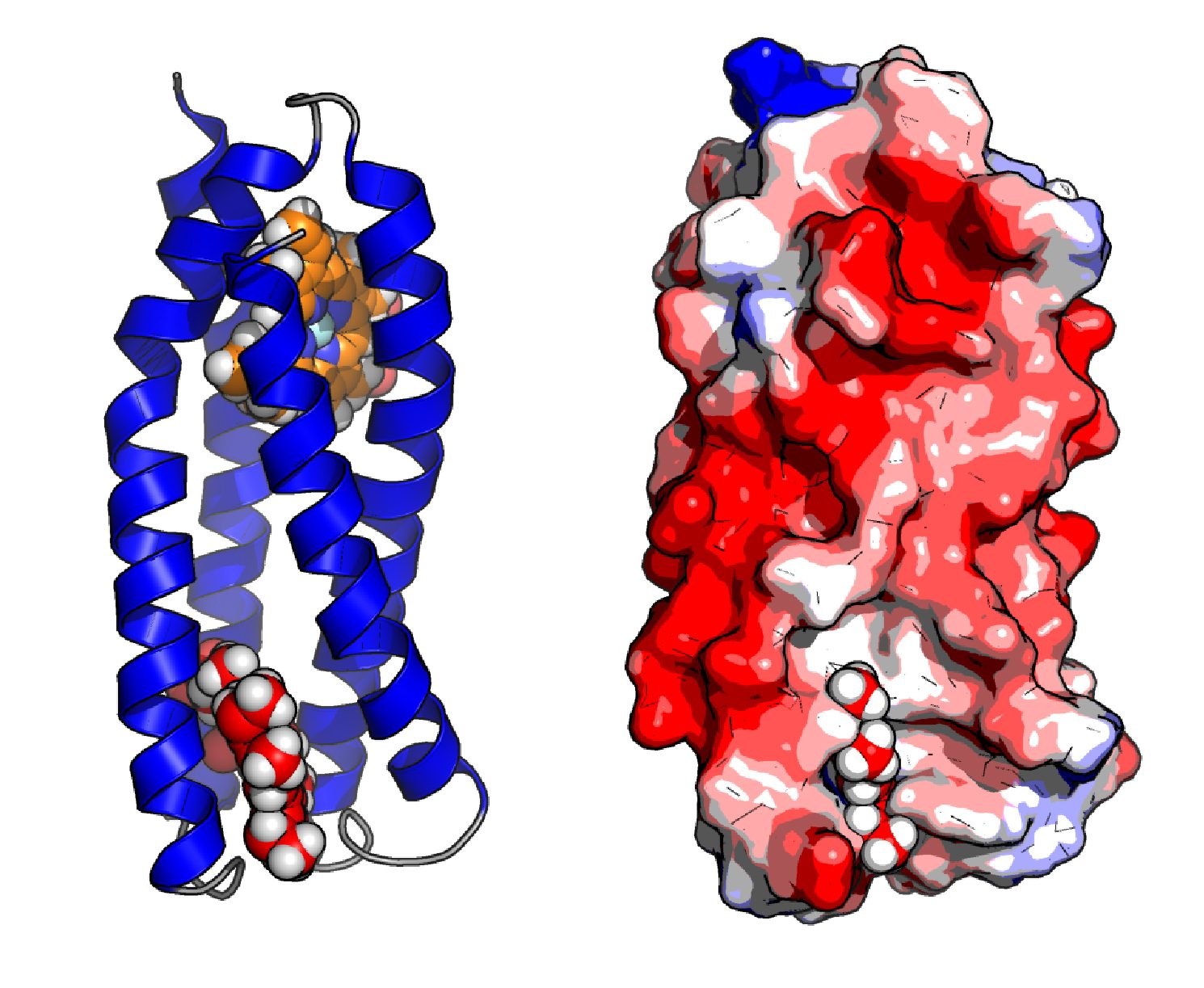
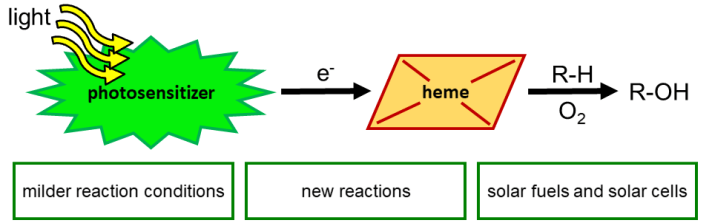
Satisfying mankind’s energy demand from sustainable sources is one of the key challenges of the 21st century. While Nature solved this challenge with photosynthesis, photosynthetic enzymes are highly unstable, which limits their biocatalytic application. We employ design and evolution to create efficient photobiocatalysts by equipping structurally simple – but catalytically proficient – heme-containing proteins with affordable organic photosensitizers. The resulting catalysts will be employed to study photon-induced electron transfer, and will be tailored for solar energy conversion and solar fuel production.
(2) Dynamic and Electrostatic Origins of Catalysis in Designer Enzymes
During my PhD (with Prof. Donald Hilvert, ETH Zurich), I have improved a computationally designed enzyme catalyzing an elementary proton transfer reaction by four orders of magnitude using directed evolution. Comparable to natural enzymes, rate enhancements were almost entirely enthalpic. Surprisingly, evolution introduced an activation heat capacity resulting in curved Eyring plots. Evolution introduced a dynamical network to recruit the whole protein for catalysis by rigidifying the TS ensemble.

Directed evolution of a related designer enzyme (Blomberg, Nature 2013) has introduced a new catalytic residue that forms an oxyanion hole. Kinetic and structural characterization of single-point variants, combined with DFT modeling, confirmed a catalytic role of oxyanion stabilization, but unexpectedly revealed that loss of that residue can be compensated by increased active-site desolvation and precise alignment of catalytic residues. We are currently using experimental and computational methods to dissect the electrostatic and dynamic origins of that effect.

Publications
Evolution of dynamical networks enhances catalysis in a designer enzyme.
Bunzel HA, Anderson JLR, Hilvert D, Arcus VL, Kamp MWvd, Mulholland AJ; Nat Chem 2021.
Rigidifying a de novo enzyme increases activity and induces a negative activation heat capacity.
Hindson SA*, Bunzel HA*, Frank B*, Svistunenko DA, Williams C, van der Kamp MW, Mulholland AJ, Pudney CR, Anderson JLR; bioRxiv 2021.
Beneficial substrate partitioning boosts non-aqueous catalysis in de novo enzyme-alginate beads.
Stenner R*, Bunzel HA*, Mulholland AJ, Anderson JLR; bioRxiv 2021.
Efficient Lewis acid catalysis of an abiological reaction in a de novo protein scaffold.
Basler S, Studer S, Zou Y, Mori T, Ota Y, Camus A, Bunzel HA, Helgeson RC, Houk KN, Jiménez-Osés G, Hilvert D; Nat Chem 2021, 13, 231.
Designing better enzymes: Insights from directed evolution.
Bunzel HA, Anderson JLR, Mulholland AJ; Curr Opin Struct Biol 2021, 67, 212.
How directed evolution reshapes the energy landscape in an enzyme to boost catalysis.
Otten R*, Pádua RAP*, Bunzel HA*, Nguyen V, Pitsawong W, Patterson M, Sui S, Perry SL, Cohen AE, Hilvert D, Kern D; Science 2020, 370, 6523, 1442-1446.
Contribution of Oxyanion Stabilization to Kemp Eliminase Efficiency.
Kries H*, Bloch JS*, Bunzel HA*, Pinkas DM, Hilvert D; ACS Catal 2020, 10, 4460.
Emergence of a negative activation heat capacity during evolution of a computationally designed enzyme
Bunzel HA, Kries H, Marchetti L, Mittl P, Mulholland A, Hilvert D; J Am Chem Soc 2019, 141, 11745.
Speeding up enzyme discovery and engineering with ultrahigh-throughput methods.
Bunzel HA*, Garrabou X*, Pott M*, Hilvert D; Curr Opin Struct Biol 2018, 48, 149.
